
What is The Deal with Editing Mitochondria?
- Ruth Wright
- Dec 16, 2020
- 5 min read

Updated: Dec 17, 2020
”The mitochondria are the powerhouse of the cell" This is what everyone ultimately remembers from school, but what is overlooked is a multitude of unique features vital to human health. Most notably, mitochondria contain their own genetic code, the mitochondrial DNA (mtDNA). The mtDNA is a double-stranded structure with approximately 16,569 base pairs which encods 37 genes. To put that into perspective, the nuclear DNA is approximately 3.2 billion base pairs long, encoding 20,000-25,000 genes. However small, these mitochondrial genes encode 13 proteins, all subunits of the oxidative phosphorylation process which is responsible for ATP synthesis and 90% of the energy required for The body to function. In the same way as the nuclear DNA can possess damaging mutation so can the mitochondrial DNA. Mutations to the mitochondrial genome can cause a large range of very severe metabolic conditions. The tissues most severely affected are those which require the most ATP; particularly the brain, skeletal and cardiac muscle but ultimately all cells require ATP to function optimally.
It should also be noted that, the nuclear DNA also possesses some genes important to mitochondrial function and as such mutations to these genes can also result in mitochondrial diseases.
Why do we want to edit the mitochondrial genome?
Mitochondrial syndromes caused by either mitochondrial or nuclear mutations affect 1 person in 4,300. These syndromes are often progressive and show a wide range symptoms and severities. Agents can be prescribed to elevate symptoms and slow disease progression but without a way of correcting the faulty DNA there is no cure.
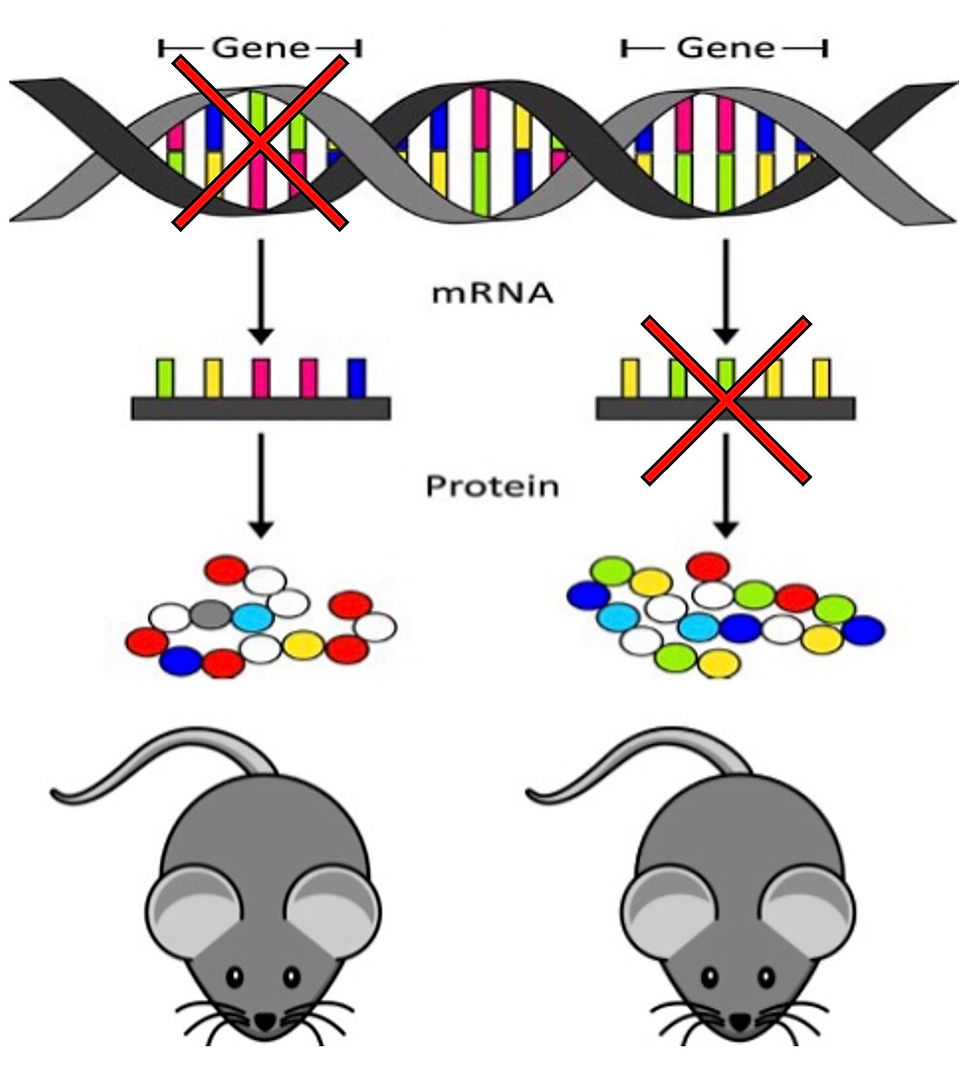
The discovery of CRISPR-Cas9 in 2011 offered hope for future cures to countless genetic diseases however, this does not include diseases caused by mutations to the mitochondrial genome.
The CRISPR-Cas9 complex consists of a guide RNA (gRNA) and a Cas9 protein. The guide RNA is designed to direct the complex to the targeted site in the genome where the Cas9 enzyme 'cuts' both strands of the DNA using cytidine deaminase. The repair machinery in the nucleus then works to repair the damage which in tern, introduces a targeted mutation. This system however is not compatible for editing the DNA contained in mitochondria. One reason for this is that the guide RNA responsible for directing the CRISPR-Cas9 complex to a target sequence is unable to pass into mitochondria.
The mitochondrial editing system
In July 2020, Mok et al. published evidence for a bacterial toxin that offers a new way of editing the mitochondrial genome (1). The bacterial toxin is another cytidine deaminase enzyme known as DddA. This enzyme is able to convert a Cytosine (C) base into a Uracil (U) base within double stranded DNA. Uracil does not naturally occur in DNA but instead is converted from Thymine (T) when RNA is transcribed from the original DNA strand. Therefore, following the conversion of C to U by DddA, the mitochondrial DNA must be replicated in order to pair the U with the correspondingAdenine (A) instead of Guanine (G) which would have been paired with C.
There are several benefits of DddA compared to the current Cas9 system. Firstly, DddA is able to act on both strands of DNA unlike the cytidine deaminase used in the CRISPR-Cas9 system which, only act on individual strands. This is significant because it removes the need of the Cas9 system which, in turn removes the need for a guide RNA to direct the Cas9 enzyme. Secondly, DddA does not 'cut' the DNA unlike the CRISPR enzyme. Instead DddA simply changes the base Cytosine to Uracil without causing the DNA damage. This is much better suited to the repair mechanisms present in the mitochondria which are far less efficient than those in the nucleus.

If the DddA enzyme were to be used on the mitochondria in its natural form it would run rampant. Every C would be converted into a U without any control. For this reason, Mok et al. divided into two halves which are harmless until brought together. Each split-DddA half is given a programmable targeting sequence that passes through the mitochondrial membranes and directs discrete DNA changes. The third and final component in this system are uracil glycosylase inhibitors attached to each half of DddA. Though the mitochondrial repair mechanisms are not as effective as those in the nucleus, they are still present and as such they must be inhibited to prevent the correction of U back to C. The inhibitors maintain the U until DNA replication can pair the U with an Adenine (A) instead of a Guanine (G). The altered DddA complex was coined DddA-derived cytosine base editor (DdCBE) which is illustrated in the figure above sources from Nature (2).
Initial analysis by Mok et al. attempted to target DdCBE to 5 mitochondrial genes within HEK293T cells. DdCBE demonstrated both an efficiency to pass into human mitochondria as well as specificity for target gene modifications. The efficiency of base pair editing ranged from 5 to 50% depending on the split type, split orientation, and target cytosine position within the spacing region of the DdCBE construct.
A major concern when working with CRISPR and other gene editing tools is the potential of DNA modifications occurring at unintended locations. Therefore, Mok and colleagues tested the possibility of DNA editing occurring following spontaneous assembly of the two DddA halves. To distinguish between DdCBE-induced changes from background differences between mitochondria within a single cell (heteroplasmy), HEK293T cells were introduced to either functional DdCBE or a corresponding dead-DdCBE control. This investigation found similar genome-wide off-target changes in both study groups which they attributed to the fact that the two halves of DdCBE must be in very close proximity to function. Another promising finding was the lack of any significant off-target changes to nuclear pseudogenes, even when the difference was only 1-2 bp from the target mitochondrial DNA. Additionally, they did not interaction in cell viability which was a point of concern with prior mitochondrial editing tools. There were also no large deletions or alterations in copy number variation.
There are a couple of limitations when using DdCBE, first being its reliance on replication. When DdCBE changes the C base to a U, the paired G on the alternate stand is left unaffected. This means when the edited gene is replicated half of the subsequent generations of DNA will have this new change and half will not, equaling a 50% effectiveness. However, to offset this DdCBE appears to actively persist for up to 18 days offering subsequent replications. Secondly, and probably most importantly, DdCBE only has the capacity to convert a C to a U which, limits its capacity. This drives the further exploration of bacterial DNA delaminates which, may expand the current toolset for mitochondrial editing.
Concluding remarks
Further investigation must also be performed to better understand how DdCbE performs on different cell types and ultimately on human cells. Regardless, this tool offers new and exciting applications for studying mitochondrial diseases using animal models and ultimately to offer future genetic therapies for those patients affected.
References:
Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, Hsu F, Radey MC, Peterson SB, Mootha VK, Mougous JD. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020 Jul;583(7817):631-7.
Aushev, M. and Herbert, M., 2020. Mitochondrial genome editing gets precise. Nature, 583(7817), pp.521-522.


Comments