
Exploration of novel mechanism for genetic compensation in the human population
- Ruth Wright
- Jan 17, 2021
- 9 min read

Advances made to genomic technologies have instigated an increase in the use of reverse genomic techniques. Model systems produced in this way have revealed profound differences between the presentations of knockout mutants and knockdown morphants.
Knockout mutants are created to have permanent gene loss of function (LoF) due to changes to the DNA sequence itself (Left Mouse).
Knockdown morphants describe organisms treated with morpholino antisense oligo nucleotides. These agents cause the temporary ‘knockdown’ of gene expression (Right Mouse).
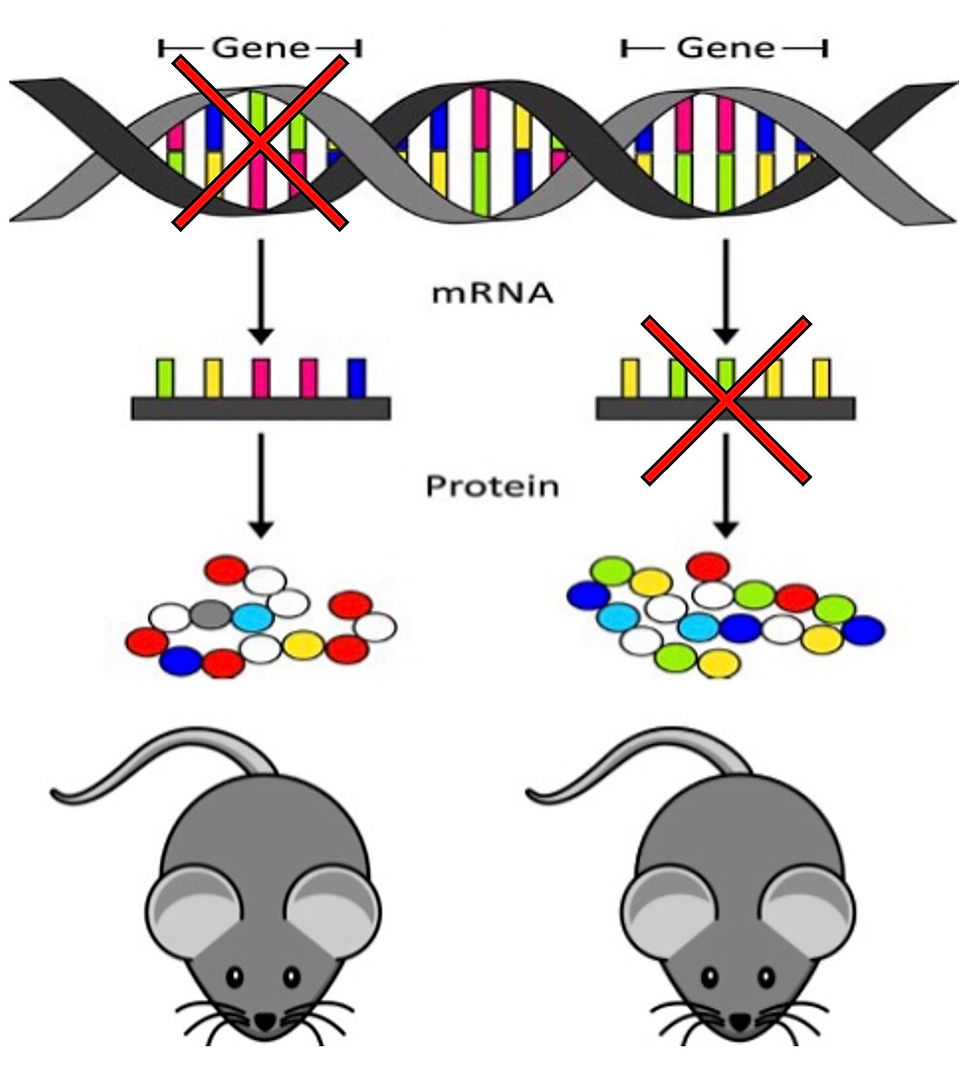
Of note, a study comparing 24 zebrafish gene mutants found ~80% of the morphant phenotypes were not observed in the corresponding mutant zebrafish (1). Findings such as these have renewed the scientific communities’ interest in a phenomenon known as genetic robustness.
Genetic robustness is a phenomenon where by an organism is able to maintain normal function despite having damaging mutations.
In general, genetic robustness is vital for species evolution as it allows an organism to survive long enough to reproduce despite minor variations in genetics. However, it also often obscures the underlying effects of gene mutations, making it difficult to study the consequence of gene LoF. To combat this, researchers studying gene mutants will often use less complex genomes such as yeast, Drosophila, rice and zebrafish as a way of minimizing the effect of genetic robustness. However, this does not completely remove the influence of robustness and thus, may still confound interpretation.
Current understanding of compensation mechanism
(You will either find this next section quite interesting or a bit dense - just a heads up)
Genetic robustness has been explained by several mechanisms including, functional redundancy of genetic pathways, requiring of genetic networks, and gene adaptation such as in highly proliferating cell populations. Recent work has described a new mechanism to explain genetic robustness, ‘transcriptional compensation’. Transcriptional compensation describes cells that functionally compensate for a knockout mutation by up-regulating the transcription of genes with a similar function to the mutated gene (2-4).
Initial studies concluded that transcriptional compensation is triggered upstream of protein function. Specifically, by mutant mRNA transcribed from a gene containing a protein truncating mutation. This is in opposition to previously identified methods for compensation which occur downstream of protein function.
A protein truncating mutation, also known as a premature termination codon (PTC), is a specific mutation that causes the premature termination of RNA production. This interferes with the subsequent protein creating an abnormally shortened protein. The effect this has on the function of the protein depends on the severity of the shortening and vital elements of the protein are lost.
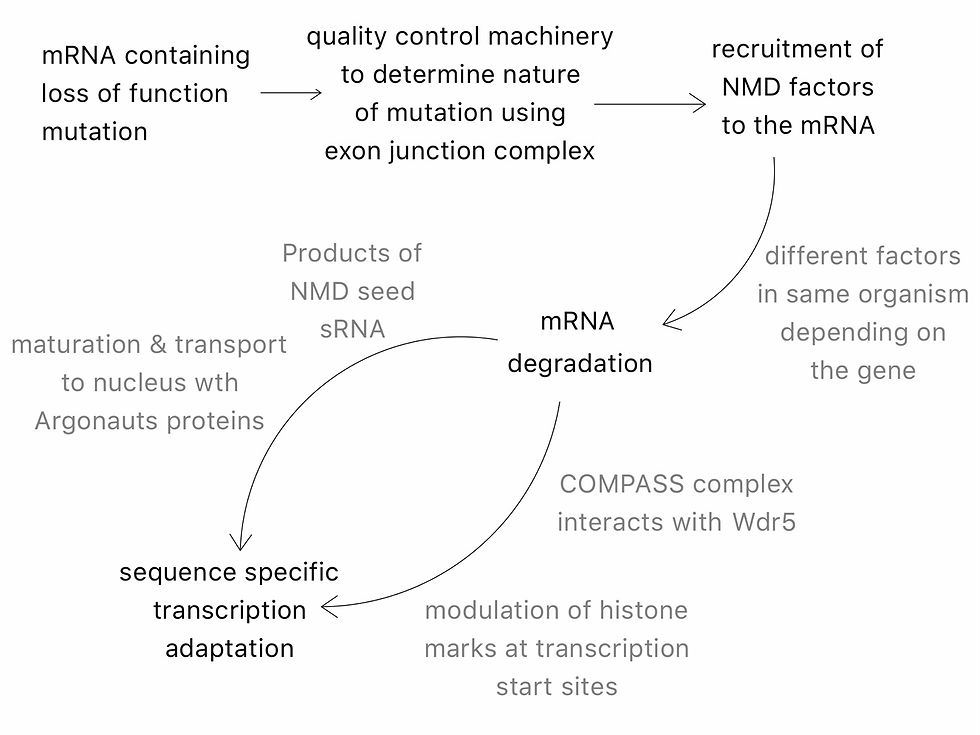
Cells protect against truncated proteins by recruiting the NMD (nonsense mediated decay) pathway. In the case of transcriptional compensation, the recruitment of NMD is thought to up-regulate functionally related genes. This explanation has been coined nonsense induced transcriptional compensation (NITC) and has most notably been explored by El-Brolosy et al. and Ma et al. (3,4).
NITC appears to selectively increase the expression of genes ancestrally related to the original PTC-bearing gene. This has been shown by the disproportionate number of the up-regulated genes which appear to have sequence similarity with the original PTC bearing gene (13). From this, it was suggested that sequence similarity may be a prerequisite for genetic compensation in zebrafish and mice (3). Moreover, even small degrees of sequence similarity have been found to result in up-regulation but, this does not always lead to functional compensation (1,3). The current, yet convoluted, understanding of NITC has been illustrated in Figure 1.
Purpose
Despite recent intrigue, the mechanisms underlying transcriptional adaptation remain poorly understood, particularly in the context of human genomics. Currently, research is limited to large-scale projects to catalogue normal and abnormal human genetic variation e.g. The Exome Aggregation Consortium. Analysis of such data has already found a surprising number of individuals possessing ‘harmful’ LoF mutations but, without any apparent symptom. This is comparable to the discrepancy observed between zebrafish morphant and mutant phenotypes. Likewise, investigations into human genetic diseases has found asymptomatic or mild forms of genetic diseases in patients possessing mutant transcripts where NMD is also present. By comparison patients without NMD develop more severe forms of the same disease (5,6). However, due to technical and ethical restrictions around engineering LoF mutations in humans have thus far prevented large-scale analysis.
Extending the study of genetic robustness to humans may also further our understanding of human knockouts, particularly in the context of drug targeting. If a human knockout exists then in theory it means that the system can tolerate the LOF of a particular gene by drug targeting. For this project, large scale genetic data was analyzed along with gene expression data to characterize NITC in the human population.
The aims of this project:
To test more broadly how transcriptional compensation acts as it remains unclear whether NITC is a property of most loss-of-function (LOF) mutations or only a handful, considering only 8 zebrafish genes and 4 mouse genes have been identified to date.
To initially establish the presence of the NITC mechanism in humans and then to characterize it.
General trends
The initial investigation focused on analyzing how tolerant a gene is to LoF versus, how similar its DNA sequence is to other genes in the genome.
LoF mutations are typically ‘damaging’ mutations and as such, usually maintain low frequencies within the human population; a consequence of natural selection. This makes assessing potential LoF mutations more challenging. Thus, LoF metrics such as pLI and LOEUF, were developed to predict how tolerant or intolerant a gene is to LoF. Should NITC be a widely used mechanism in humans then, a correlation should be observed between higher LoF tolerance (i.e. low pLI and high LOEUF) and high sequence similarity (measured as cross mappability).
high pLI (≥0.9) or low LOEUF decile indicates extreme intolerance to LoF where as, low pLI (≤0.1) or high LOEUF decile indicates LoF tolerance.
To test whether genes that are more tolerant to LoF mutations show increased sequence similarity compared to other regions of the genome, genes were split into extremely intolerant or tolerant groupings. A comparison was then made between the degrees of cross mappability observed in each of the groups.
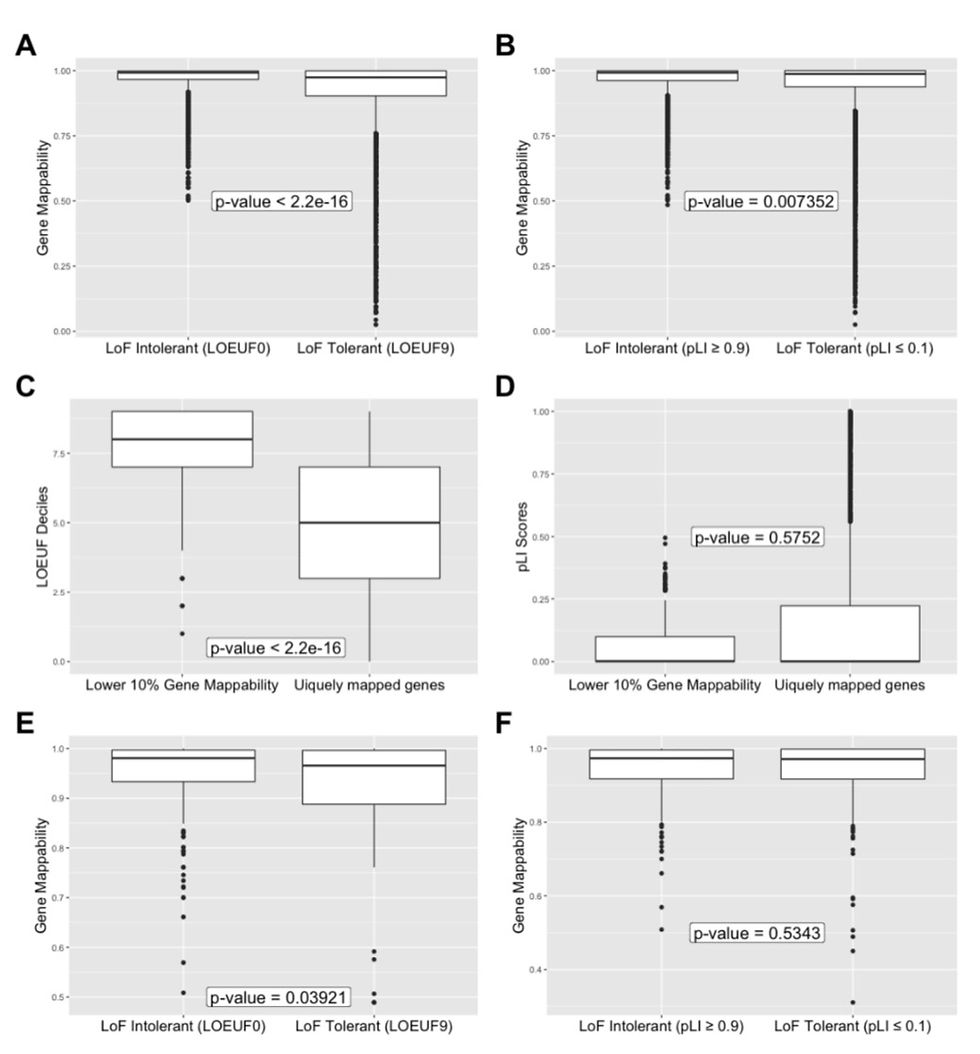
The findings suggested that genes with more LoF tolerance show significantly higher redundancy in the human genome with, more regions of sequence similarity and by extension functional similarity. Refer to figures A and B for results using LOEUF and pLI, respectively. This effect was also found when data was filtered for only essential genes (figures E and F).
Compared to non-essential genes, essential genes experience a greater degree of constraint by natural selection. Thus, are typically more intolerant to LoF mutations (7). That being the case, essential genes with LoF tolerance must have to have some method by which they compensate for the loss of gene function and therefore, maintain normal system function. This was supported by the slightly larger effect size observed in the essential genes versus the unfiltered genes when using the LOEUF metric.
What is essential?
It should be noted that in defining ‘essential’ and ‘nonessential’ genes, transcriptionally compensated genes may evade investigation and thus characterization for essentiality. Transcriptionally compensated genes may in fact show the greatest degree of compensation which, may explain why no significant or suggestive gene pairs matched with known human essential genes. When assessing human genes for essentiality the two most common methods are CRISPR/Cas9 screens and gene-trap vector screens (8). The degree to which current methods for identifying essential human genes may be affected by compensation would be an interesting avenue for further investigation.
Results for the analysis with LOEUF and pLI metrics were largely comparable except for an insignificant result obtained from part of the pLI analysis. PLI scores however, are limited to only characterizing probable LoF variants’ in small genes (7). This is due to the fact that smaller genes are naturally more difficult to assess for LoF intolerance as they contain a far fewer nucleotide bases, meaning that the number of expected over observed degrees of tolerance is also reduced. The LOEUF score improves upon this but Karczewski et al. note that a high LOEUF decile will contain a mix of genes without LoF depletion as well as small genes (7).
This however does not present as large a problem in this specific analysis. This is because these high LOEUF genes define LoF tolerant genes and are the focus of this analysis. Meaning this analysis focused on low LOEUF genes and therefore excludes small genes. By comparison, when assessing deleterious LoF mutations for pathogenicity the focus would be on LoF intolerance and therefore the previously stated limitations of these metrics need to be concidered.
NITC in RNA Sequencing Data
Analysis of individual gene expression levels from various different tissue types found evidence of LoF mutations causing changes in expression of genes with similar sequence to gene containing LoF mutation. The only statistically significant finding was between a splice acceptor variant in PSG9, which was associated with a decrease in expression of PSG5. On top of this, the variant is not located in the last exon of PSG9 meaning, it is not exempt from NMD. This supports the idea that the compensatory mechanism at work may be NITC.
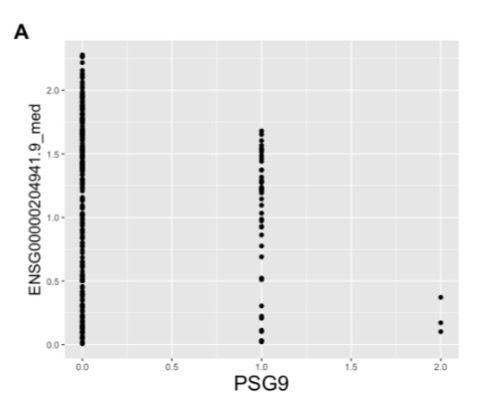
Both PSG9 and PSG5 are members of the pregnancy-specific glycoprotein family, part of the larger immunoglobin superfamily. Reduced serum concentration of the PSG family has been associated with decreased foetal growth and more specifically, the identified PSG9 variant (rs3746297) has been shown to be associated with recurrent pregnancy loss when, combined with other gene polymorphisms or blood coagulation factors (9).
Lowering the threshold of significance
When the threshold for statistical significance was lowered to encompass ‘suggestive’ associations a total of 17 associations (not including the PSG9 variant) were identified, all of which were mutations located in either IFIT5 or C2orf91. These associations had a wide range of cross mappability scores meaning various degrees of gene similarity. The variants with very low gene similarity are not of great concern as there is evidence to suggest that even genes with a limited degree of sequence similarity can experience up-regulation during NTIC (3).
Future work
Additional datasets
The observations made in this research project provide some evidence for NITC being a mechanism present in the human system. However, it is based on trans-eQTL data which have several innate limitations.
Due to limited power, con founders, and small effect sizes it has proven difficult to identify trans-eQTLs in human data. Another challenge with trans-eQTL analysis stems from the fact that the test is genome wide. Consequently, is more prone to systemic errors when compared to cis-eQTL analysis. As such, observations presented in this research should also be validated with analysis of further gene expression cohorts such as, CartAGene and Expression Atlas. These datasets would allow for greater generalizability of the results while validating any significant or suggestive gene pairs identified.
Read mapping re-analysis
Previous studies have reported that significant distant eQTL results may be driven by artefacts arising from mapping and alignment errors (10). For the results detailed here, it should be noted that genes with altered expression associated to a LoF mutation are similar in sequence to the LoF mutation bearing gene. A potential consequence of this is that reads that map to both the LoF bearing gene and associated gene may have been discarded during initial quality control measures. It would therefore be advisable to repeat this analysis using the tool 'RNA-Seq by expectation maximization' (RSEM) to qualify expression.
RSEM assigns ambiguous reads to a given gene/transcript instead of simply filtering them out entirely. Comparison between the results obtained here and when quantifying expression with RSEM may elucidate the proportion of reads removed for ambiguity.
Biological studies
Contrary to what is expected for NITC which, describes an up-regulation of gene expression, modulation of the PSG5 warrants further study in alternate tissues types and disease settings. Additionally, an important drawback to the PSG9 PSG5 association is the fact that it was identified in transformed fibroblast tissue samples. This is contrary to the RNA tissue specificity of PSG9 for placental tissue and furthermore its function in foetal development. Where current literature stands, there is evidence for paralogues and related genes to show expression at discrete times and in discrete tissues (11). However, it is unclear whether adapting gene expression can appear in either tissues or at times where they are not typically expressed. As such, future work should focus on validating these observations and developing stronger biological inferences.
Genomic context
Due to a restricted timeline, an in-depth characterization of this genetic compensation mechanism was unattainable. This would be a recommended point of inquiry for future work.
Analyses may include an investigation of the genomic context surrounding the LoF mutations, which may include regulatory marks such as enhancers, histone marks, methylation patterns, transcription factors, and motifs. Additionally, the length of the truncated transcript could be tested for possible influence on the specific compensatory response.
Conclusion
In summary, the results obtained from this analysis present a case for further investigation into nonsense induced transcriptional compensation as an explanation for genetic robustness observed in the human population.
References
Rossi A, Kontarakis Z, Gerri C, Nolte H, Hölper S, Krüger M et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524(7564):230-233.
El-Brolosy M, Stainier D. Genetic compensation: A phenomenon in search of mechanisms. PLOS Genetics. 2017;13(7):e1006780.
El-Brolosy, M.A., Kontarakis, Z., Rossi, A., Kuenne, C., Günther, S., Fukuda, N., Kikhi, K., Boezio, G.L.M., Takacs, C.M., Lai, S.-L., et al. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197.
Ma, Z., Zhu, P., Shi, H., Guo, L., Zhang, Q., Chen, Y., Chen, S., Zhang, Z., Peng, J., and Chen, J. (2019). PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568, 259–263.
Dietz, H. C. et al. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics 17, 468-475, doi:10.1006/geno.1993.1349 (1993).
Hall, G. W. & Thein, S. Nonsense codon mutations in the terminal exon of the beta-globin gene are not associated with a reduction in beta-mRNA accumulation: a mechanism for the phenotype of dominant beta-thalassemia. Blood 83, 2031-2037 (1994).
Karczewski K, Francioli L, Tiao G, Cummings B, Alföldi J, Wang Q et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434-443.
Fraser A. Essential Human Genes. Cell Systems. 2015;1(6):381-382.
Lee H, Ahn E, Kim J, Kim J, Ryu C, Lee J et al. Association study of frameshift and splice variant polymorphisms with risk of idiopathic recurrent pregnancy loss. Molecular Medicine Reports. 2018;.
Pickrell JK, Marioni JC, Pai AA, et al.: Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010; 464(7289): 768–772
Jojic B, Amodeo S, Bregy I, Ochsenreiter T. Distinct 3′ UTRs regulate the life- cycle-specific expression of twoTCTPparalogs inTrypanosoma brucei. Journal of Cell Science. 2018;131(9):jcs206417.
Peng J. Gene redundancy and gene compensation: An updated view. Journal of Genetics and Genomics. 2019;46(7):329-333.


Comments